|
Поделиться ссылкой:
Прочитать статью в pdf формате
Биология желчных кислот
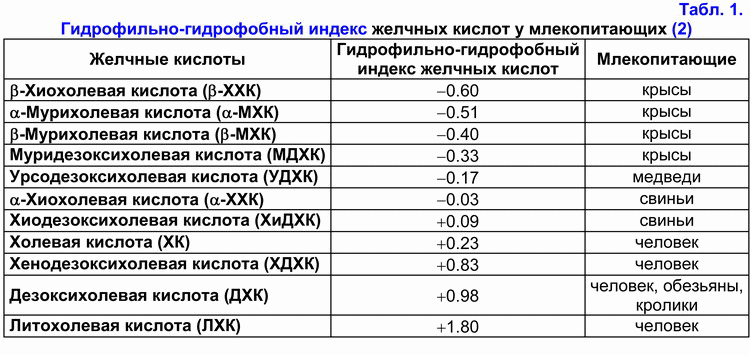
На основании гидрофильно-гидрофобного индекса желчные кислоты подразделяют на гидрофильные и гидрофобные (табл. 1) (1-3).
Если гидрофильно-гидрофобный индекс меньше гидрофильно-гидрофобного индекса холевой кислоты (ХК), то эти желчные кислоты относят к гидрофильным, если больше – то к гидрофобным (1-3). Первичные желчные кислоты более гидрофильные, чем вторичные, а тауриновые коньюгаты желчных кислот более гидрофильные, чем глициновые (1-3). Гидрофильные желчные кислоты обладают гепатозащитными свойствами (мурихолевая (МХК) > урсодезоксихолевая (УДХК) > ХК) (4, 5). Гидрофобные желчные кислоты являются гепатотоксичными (литохолевая (ЛХК) > дезоксихолевая (ДХК) > хенодезоксихолевая (ХДХК) > ХК) (1-7). В зависимости от концентрации они вызывают холестаз (ЛХК > ДХК), некроз (ЛХК > ДХК) или апоптоз гепатоцитов (ЛХК > ДХК > ХДХК) (2-7). ДХК к тому же обладает канцерогенными свойствами (8). В эксперименте на животных продемонстрировано, что она вызывает рак толстой кишки (9). Гидрофильные желчные кислоты предупреждают развитие холестаза или некроза/апоптоза гепатоцитов (УДХК, МХК), а также рак толстой кишки (УДХК) (4-7, 9).
В сыворотке крови до 40% желчных кислот транспортируется ЛПВП, до 15% - с ЛПНП (10). Механизм связывания желчных кислот с липопротеидами зависит от их гидрофильно-гидрофобного индекса (ХДХК > ДХК > УДХК > ХК > 7-эпихолевая кислота) (10). В печени 60-80% желчных кислот захватываются за один проход портальной крови (11). Ранее в экспериментах на хомяках было показано, что печеночный захват ЛПНП может влиять на скорость секреции желчи, желчных кислот и холестерина (12, 13). Состав и концентрация желчных кислот, участвующих в энтерогепатической циркуляции, может модулировать действие ЛПНП рецепторов и рецептор-зависимый захват ЛПНП в печени. Более гидрофильная УДХК стимулирует рецептор-зависимый захват ЛПНП в печени, а более гидрофобная ХДХК - снижает активность ЛПНП рецепторов (12, 13). Также было показано, что добавление гидрофобной ХДХК к гиперхолестериновой диете уменьшает концентрацию ЛПВП в сыворотке крови, а добавление гидрофильной УДХК вызывает обратную картину (14, 15). В гепатоцитах желчные кислоты могут ингибировать активность ГМГ-КоА-редуктазы и холестерин-7a-гидроксилазы в зависимости от их концентрации и гидрофильно-гидрофобного индекса (ДХК > ХДХК > ХК > УДХК) (2, 16-18). Гидрофильные желчные кислоты стимулируют секрецию печеночной желчи (УДХК > ХК), гидрофобные - снижают (ЛХ > ДХК > ХДХК) (19-21). УДХК и ХДХК уменьшают секрецию билиарного холестерина в печеночной желчи, ХК и ДХК – повышают (1, 19-21). В пузырной желчи гидрофобные желчные кислоты формируют смешанные (желчная кислота-фосфолипид-холестерин) и простые (желчная кислота-холестерин) мицеллы (ДХК > ХДХК > ХК), а гидрофильные желчные кислоты – жидкокристаллические ламеллы (МХК > УДХК) (22-25). Т.е. чем меньше гидрофильно-гидрофобный индекс желчных кислот, тем ниже их способность формировать мицеллы. В подвздошной кишке ХК и ХДХК повышают абсорбцию холестерина, а УДХК и ДХК – снижают (26-29). В процессе энтерогепатической циркуляции в кишечнике под воздействием анаэробных бактерий происходит 7a-дегидроксилирование первичных желчных кислот (хиохолевой (ХХК), МХК, ХК, ХДХК) и образование вторичных желчных кислот (хиодезоксихолевой (ХиДХК), муридезоксихолевой (МДХ), ДХК, ЛХК) (1, 2, 30, 31). Вторичные желчные кислоты более гидрофобны, чем первичные (ХиДХК > ХХК, МДХ > МХК, ДХК > ХК, ЛХК > ХДХК) (1-3). В норме вторичные желчные кислоты плохо всасываются в подвздошной и толстой кишке и выделяются с фекалиями (1-3).
Механизм формирования литогенной желчи
Ранее нами была показана повышенная экспрессия циклооксигеназы 2 (ЦОГ-2) в стенке желчного пузыря (ЖП), полученных после холецистэктомии от больных хроническим калькулезным холециститом (ХКХ) (n=21), в гладкомышечных клетках - 86%, в эпителиальных клетках - 81%, в стенках сосудов - 71%, в стромальных клетках - 57%, в синусах Рокитанского-Ашоффа - 37% (32). При выраженности интенсивности воспаления в стенке желчного пузыря слабой степени (n=12) повышенная экспрессия ЦОГ-2 обнаружена в эпителиальных клетках - 83%, в стенке сосудов - 78%, в гладкомышечных клетках - 75%, в стромальных клетках - 33%, в синусах Рокитанского-Ашоффа - 17%. В группе, включающей более выраженную степень воспаления (умеренную и резкую, n=9), повышенная экспрессия ЦОГ-2 была определена в гладкомышечных клетках - 100%, в стенках сосудов - 89%, в эпителиальных клетках - 78%, в стромальных клетках - 78%, в синусах Рокитанского-Ашоффа - 67%. Выявлена положительная корреляция между выраженностью воспаления в стенке желчного пузыря и выраженностью экспрессии ЦОГ-2 в гладкомышечных клетках (r= +0.71, p<0.001) и стенках сосудов (r= +0.51, p<0.05). При исследовании желчных пузырей без метаплазии (n=13) повышенная экспрессия ЦОГ-2 обнаружена в гладкомышечных клетках - 85%, в эпителиальных клетках - 69%, в стенках сосудов - 69%, в стромальных клетках - 54%, в синусах Рокитанского-Ашоффа - 38%. Интенсивность воспаления в стенке желчного пузыря зависела от экспрессии ЦОГ-2 в гладкомышечных клетках (r= +0.82, p<0.001). У больных ХКХ выявлена отрицательная корреляция между абсорбционной функцией желчного пузыря и толщиной стенки желчного пузыря (r= -0.71, p<0.05) (33).
Полученные данные свидетельствуют:
- Повышенная экспрессия ЦОГ-2 в гладкомышечных клетках, стенках сосудов и эпителиальных клетках желчного пузыря может быть причиной хронического асептического воспаления, снижения абсорбции воды и “пассивного” пассажа печеночной желчи в желчный пузырь до 35%.
- Избыточная экспрессия ЦОГ-2 в гладкомышечных клетках может быть причиной гипомоторной дисфункции желчного пузыря и болевого синдрома.
- Избыточная экспрессия ЦОГ-2 в гладкомышечных клетках, стенках сосудов и эпителиальных клетках желчного пузыря может быть причиной увеличения толщины стенки желчного пузыря.
- Избыточная экспрессия ЦОГ-2 в эпителиальных клетках желчного пузыря может быть причиной гиперсекреции гликопротеинового муцина в просвет желчного пузыря и повышения концентрации гликопротеинового муцина в пузырной желчи.
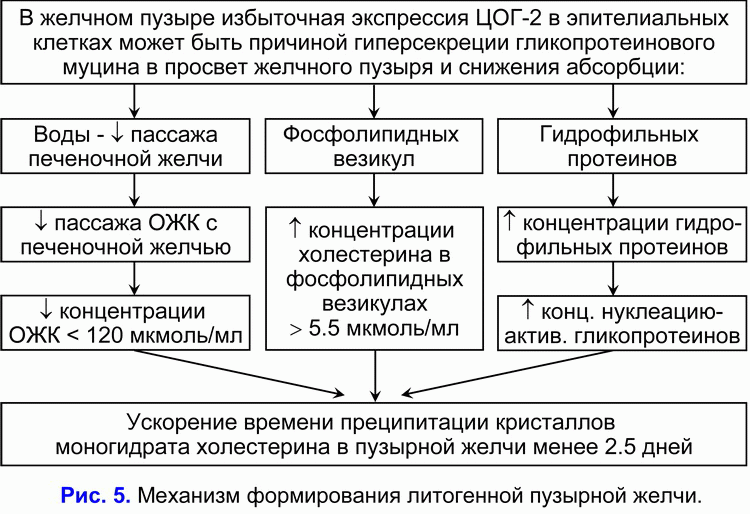
Принимая во внимание, что повышенная экспрессия ЦОГ-2 в гладкомышечных клетках, стенках сосудов и эпителиальных клетках желчного пузыря может проявляться на ранних этапах холецистолитиаза, то повышенная экспрессия ЦОГ-2 в гладкомышечных клетках, стенках сосудов и эпителиальных клетках желчного пузыря может быть физической причиной формирования хронического внутрипузырного холестаза и “литогенной” пузырной желчи:
1) уменьшать абсорбцию воды слизистой желчного пузыря и обуславливать снижение скорости поступления желчных кислот печеночной желчи в желчный пузырь (ограничение “пассивного” пассажа) и концентрации общих желчных кислот в пузырной желчи;
2) снижать абсорбцию везикулярного холестерина слизистой желчного пузыря и способствовать увеличению концентрации холестерина в фосфолипидных везикулах в пузырной желчи;
3) снижать абсорбцию гидрофильных протеинов слизистой желчного пузыря и повышать их концентрацию в пузырной желчи.
Это сопровождается увеличением соотношения везикулярный холестерин/общие желчные кислоты и общие протеины/общие желчные кислоты и способствует росту скорости преципитации кристаллов моногидрата холестерина на эпителиальных клетках слизистой желчного пузыря. Следовательно, чем меньше скорость абсорбции везикулярного холестерина слизистой желчного пузыря, тем больше его в пузырной желчи, и меньше время нуклеации кристаллов моногидрата холестерина в пузырной желчи, и наоборот. Таким образом, повышенная экспрессия ЦОГ-2 эпителиальных клетках желчного пузыря, снижая абсорбционную и концентрационную функцию желчного пузыря, способствует формированию “литогенной” пузырной желчи. Уменьшение моторно-эвакуаторной функции желчного пузыря (повышенная экспрессия ЦОГ-2 в гладкомышечных клетках желчного пузыря) является предрасполагающим фактором для формирования желчных камней (рис 5).
Уменьшение поступления печеночной желчи в желчный пузырь повышает ее выделение в двенадцатиперстную кишку, увеличивает количество циклов пузырно-независимой энтерогепатической циркуляции желчных кислот и стимулирует образование гидрофобной гепатотоксичной дезоксихолевой желчной кислоты (ДХК) (10, 34, 35).
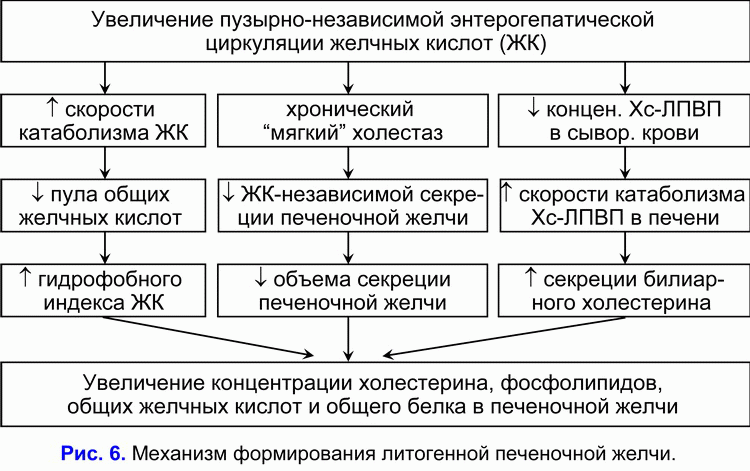
Увеличение циклов пузырно-независимой энтерогепатической циркуляции желчных кислот и концентрации гидрофобной гепатотоксичной дезоксихолевой желчной кислоты в гепатоцитах снижает желчно-кислото-независимый тип секреции печеночной желчи и стимулирует формирование хронического “мягкого” внутрипеченочного холестаза (36, 37). Таким образом, снижение пассажа печеночной желчи в желчный пузырь и, соответственно, увеличение пассажа печеночной желчи в двенадцатиперстную кишку является причиной повышения частоты циклов пузырно-независимой энтерогепатической циркуляции желчных кислот и возникновения хронического “мягкого” внутрипеченочного холестаза.
Хронический “мягкий” внутрипеченочный холестаз характеризуется снижением объема секреции печеночной желчи и повышением в ней концентрации холестерина, общих желчных кислот и общих протеинов (рис. 6) (38, 39). Увеличение концентрации холестерина в печеночной желчи способствует повышению содержания холестерина в фосфолипидных везикулах (r= +0.59, p<0.05) (40). Повышение уровня общих желчных кислот в печеночной желчи снижает стабильность фосфолипидных везикул и укорачивает время нуклеации кристаллов моногидрата холестерина (r= -0.53, p<0.05) (40). Мы полагаем, что хронический “мягкий” внутрипеченочный холестаз, снижая скорость секреции и объем печеночной желчи, способствует повышению концентрации холестерина, общих желчных кислот, протеинов и уменьшению времени нуклеации кристаллов моногидрата холестерина, т.е. формированию литогенной печеночной желчи.
Снижение абсорбционной, концентрационной и эвакуаторной функций желчного пузыря способствует формированию литогенной пузырной желчи, хронический “мягкий” внутрипеченочный холестаз - литогенной печеночной желчи (рис 5, рис 6). Эти два фактора определяют формирование холестериновых желчных камней.
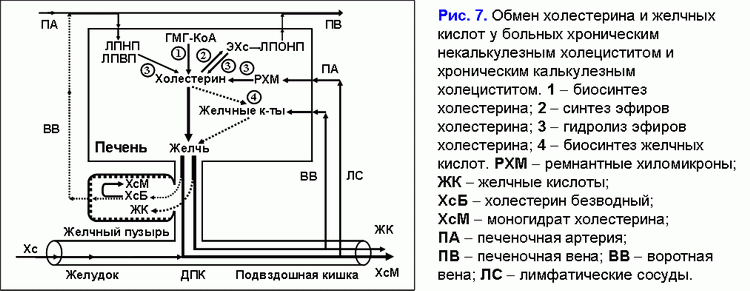
У больных хроническим некалькулезным холециститом с билиарным сладжем снижение абсорбционной (снижение абсорбции воды и фосфолипидных везикул), концентрационной (снижение концентрации общих желчных кислот в пузырной желчи) и эвакуаторной функций (снижение пузырно-зависимого выхода билиарного холестерина) и увеличение секреторной функции (гиперсекреция гликопротеинового муцина слизистой) желчного пузыря способствует образованию холестериновых желчных камней (рис. 7) (41).
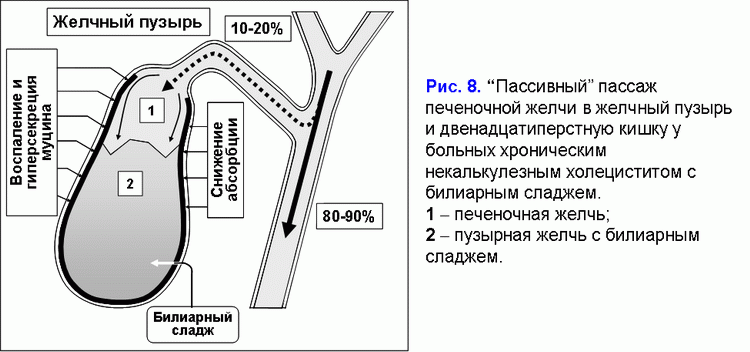
Уменьшение абсорбции воды в стенке желчного пузыря ограничивает “пассивный” пассаж печеночной желчи в желчный пузырь и увеличивает - в двенадцатиперстную кишку (рис. 8) (41-43).
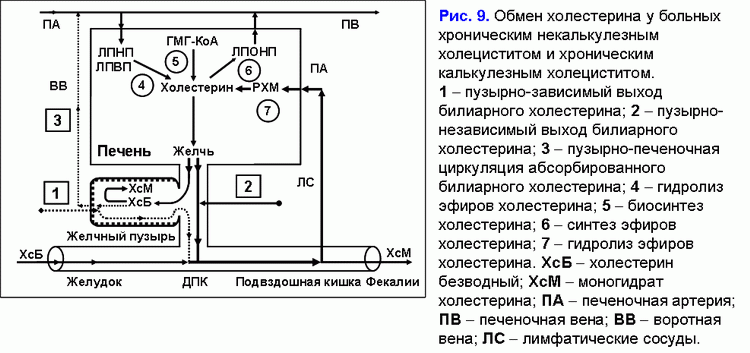
Снижение эвакуаторной функции желчного пузыря уменьшает “активный” пассаж печеночной желчи в желчный пузырь (44, 45). Это сопровождается снижением концентрации общих желчных кислот и увеличением концентрации билиарного холестерина в фосфолипидных везикулах и способствует увеличению времени для преципитации кристаллов моногидрата холестерина и формирования холестериновых желчных камней (рис. 9) (46-50).
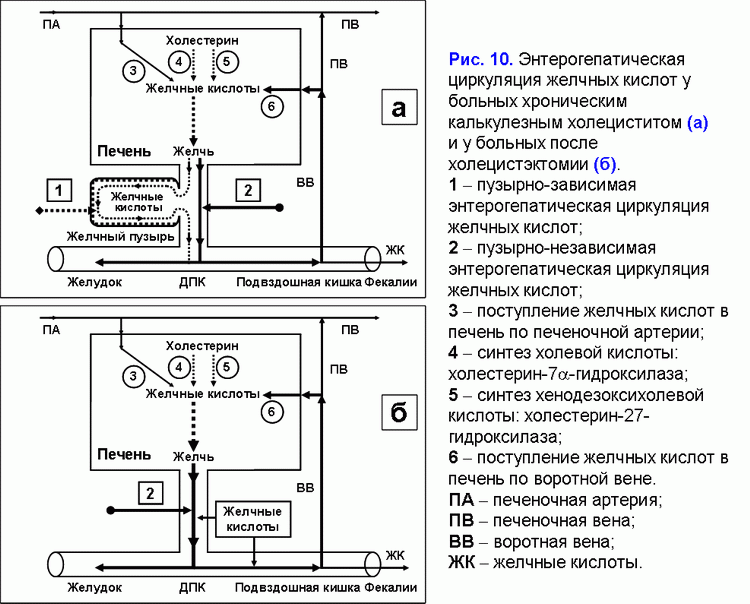
Избыточный пассаж печеночной желчи в двенадцатиперстную кишку увеличивает частоту пузырно-независимой энтерогепатической циркуляции желчных кислот. У больных хроническим калькулезным холециститом или после холецистэктомии повышена пузырно-независимая энтерогепатическая циркуляция желчных кислот (рис. 10).
Как следствие у них увеличено образование гидрофобной гепатотоксичной дезоксихолевой желчной кислоты (табл. 3) и накопление ее в гепатоцитах (51), формирование морфологических изменений в печени (неспецифический реактивный гепатит) (52) и возникновение холестаза (53), а также повышается риск рака поджелудочной железы и печени, толстой и тонкой кишки (54-62). Увеличение ДХК, участвующей в энтерогепатической циркуляции, и других токсических веществ в печеночной желчи может поддерживать появление хронического панкреатита, дуодено-гастрального рефлюкса (63-66).
Список литературы:
- Hofmann A.F. Bile secretion and the enterohepatic circulation of bile acids. In: Feldman M., Scharschmidt B.F., Sleisenger M.H., eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management. 6th ed. Philadelphia: WB Saunders Company, 1998: 937-948.
- Heuman D.M., Hylemon P.B., Vlahcevic Z.R. Regulation of bile acid synthesis. III. Correlation between biliary bile acid hydrophobicity index and the activities of enzymes regulating cholesterol and bile acid synthesis in the rat. J Lipid Res 1989; 30: 1161-1171.
- Hofmann A.F. Bile Acids. In: Arias I.M., Boyer J.L., Fausto N., Jakoby W.B., Schachter D.A., Shafritz D.A., eds. The Liver, Biology and Pathobiology. 3rd ed. New York: Raven Press, 1994: 677-718.
- Roda A., Piazza F., Baraldini M., Speconi E., Guerra M.C., Cerre C., Forti G.C. Taurohyodeoxycholic acid protects against taurochenodeoxycholic acid-induced cholestasis in the rat. Hepatology 1998; 27: 520-525.
- Scholmerich J., Baumgartner U., Miyai K., Gerok W. Tauroursodeoxycholate prevents taurolithocholate-induced cholestasis and toxicity in rat liver. J Hepatol 1990; 10(3): 280-283.
- Lim A.G., Ahmed H.A., Jazrawi R.P., Levy J.H., Northfield T.C. Effects of bile acids on human hepatic mitochondria. Eur J Gastroenterol Hepatol 1994; 6(12): 1157-1163.
- Paumgartner G., Beuers U. Bile acids and the liver. In: Surrenti C., Casini A., Milani S., Pinzani M., eds. Fat-storing Cells and Liver Fibrosis. Dordrecht: Kluwer Academic Publishers, 1994: 330-339.
- Ochsenkuhn T; Bayerderffer E; Meining A; Schinkel M; Thiede C; Nussler V; Sackmann M; Hatz R; Neubauer A; Paumgartner G. Colonic mucosal proliferation is related to serum deoxycholic acid levels. Cancer 1999; 85(8): 1664-1669.
- Shekels L.L., Beste J.E., Ho S.B. Tauroursodeoxycholic acid protects in vitro models of human colonic cancer cells from cytotoxic effects of hydrophobic bile acids. Lab Clin Med 1996; 127: 57-66.
- Carey M.C., Duane W.C. Enterohepatic circulation. In: Arias I.M., Boyer J.L., Fausto N., Jakoby W.B., Schachter D.A., Shafritz D.A., eds. The Liver, Biology and Pathobiology. 3rd ed. New York: Raven Press, 1994: 719-767.
- Ewerth S., Angelin B., Einarsson K., Nilsell K., Bjorkhem I. Serum concentrations of ursodeoxycholic acid in portal venous and systemic venous blood of fasting humans as determined by isotope dilution-mass spectrometry. Gastroenterology 1985; 88:126-133.
- Malavolti M., Ceryak S., Fromm H. Modulation of bile secretion by hepatic low-density lipoprotein uptake and by chenodeoxycholic acid and ursodeoxycholic acid treatment in the hamster. Gastroenterology 1987; 93: 1104-1115.
- Malavolti M., Fromm H., Ceryak S., Roberts I.M. Modulation of low-density lipoprotein receptor activity by bile acids: differential effects of chenodeoxycholic and ursodeoxycholic acids in hamster. J Lipid Res 1987; 28: 1281-1295.
- Ceryak S., Bouscarel B., Malavolti M., Robins S.J., Fromm H. Effect of ursodeoxycholic bile acid on hepatic LDL metabolism in dietary hypercholesterolemic hamsters. Gastroenterology 1996; 110: A1165.
- Fromm H., Bouscarel B., Ceryak S., Malavolti M. Direct effects of bile acids on low-density lipoprotein metabolism. In: Fromm H., Leuschner U., eds. Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 141-144.
- Maton P.N., Ellis H.J., Higgins J.P., Dowling R.H. Hepatic HMG-CoA reductase in human cholelithiasis: effect of chenodeoxycholic and ursodeoxycholic acids. Europ J Clin Invest 1980; 10: 325-332.
- Vlahcevic Z.R., Heuman D.H., Hylemon P.B. Regulation of bile acid synthesis. Hepatology 1991; 13: 590-600.
- Vlahcevic Z.R. Regulation of cholesterol 7a-hydroxylase by different effectors. Ital J Gastroenterol 1996; 28: 337-339.
- Lindbland L., Lundholm K., Schersten T. Influence of cholic and chenodeoxycholic acid on biliary cholesterol secretion in man. Europ J Clin Invest 1977; 7: 383-388.
- Sama C., LaRusso N.F., Loper del Pino V., Thistle J.L. Effects of acute bile acid administration on biliary lipid secretion in healthy volunteers. Gastroenterology 1982; 82: 515-525.
- Carulli N., Loria P., Bertolotti M. Effects of acute change of bile acid pool composition on biliary lipid secretion. J Clin Invest 1984; 74: 614-624.
- Carey M.C. Pathogenesis of gallstones. Amer J Surg 1993; 165: 410-419.
- Carey M.C. Formation and growth of cholesterol gallstones: the new synthesis. In: Fromm H., Leuschner U., eds. Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 147-175.
- Lichtenberg D., Ragimova S., Bor A., Almog S., Vinkler C., Peled Y., Halpern Z. Stability of mixed micellar systems made by solubilizing phosphatidylcholine-cholesterol vesicles by bile salts. Hepatology 1990; 12: 149S-154S.
- Van de Heijning B.J.M., Stolk M.F.J., van Erpecum K.J., Renooij W., van Berge Henegouwen G.P. The effects of bile salt hydrophobicity on model bile vesicles morphology. Biochim Biophys Acta 1994; 1212: 203-210.
- Einarsson K., Grundy S.M. Effects of feeding cholic and chenodeoxycholic acid on cholesterol absorption and hepatic secretion of biliary lipids in man. J Lipid Res 1980; 21: 23-34.
- Ponz de Leon M., Carulli N. The influence of bile acid pool composition on the regulation of cholesterol absorption. In: Paumgartner G., Stiehl A., Gerok W., eds. Bile Acids and Lipids. London: MTP Press, 1981: 133-140.
- Sama C., LaRusso N.F. Effect of deoxycholic, chenodeoxycholic, and cholic acids on intestinal absorption in humans. Mayo Clin Proc 1982; 57: 44-50.
- Lanzini A., Northfield T.C. Effect of ursodeoxycholic acid on biliary lipid coupling and on cholesterol absorption during fasting and eating in subjects with cholesterol gallstones. Gastroenterology 1988; 95(2): 408-416.
- Christl S.U., Bartram H.P., Paul A., Kelber E., Scheppach W., Kasper H. Bile acid metabolism by colonic bacteria in continous culture: effects of strach and pH. Ann. Nutrition metabolism 1997; 41: 45-51.
- Farkkila M.A., Turunen V.M., Miettinen T.A. The role of intestinal bacteria in regulation of cholesterol metabolism. Gastroenterology 1994; 106(4): A891.
- Golubev S.S., Kozlova N.M., Turumin J.L., Raevskaja L.Ju. The expression of cyclooxygenase-2 in the gallbladder wall of the patients with chronic calculous cholecystitis. Acta Gastroenterologica Belgica 2007; 70(1): Abstr. 31.
- Kozlova N.M., Turumin J.L., Galeev Y.M., Popov M.V. The functional state of hepato-biliary system in the patients with chronic calculous cholecystitis in the stage of exacerbation. Acta Gastroenterologica Belgica 2007; 70(1): Abstr. 33.
- Roda E. Changes of biliary lipid secretion and bile acid pool size in pathogenesis of cholesterol gallstones. Falk Symposium No. 84: Bile Acids-Cholestasis-Gallstones - Advances in Basic and Clinical Bile Acid Research: Abstr. Book. Berlin, Germany, 1995: Abstr. 26.
- Roda E., Cipolla A., Bazzoli F. et al. Modifications in biliary lipid secretion and bile acid kinetics in the pathogenesis of cholesterol gallstones. In: H. Fromm, U. Leuschner, eds Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 176-179.
- Honda A., Yoshida T., Tanaka N. et al. Increased bile acid concentration in liver tissue with cholesterol gallstone disease. J. Gastroenterol. 1995; 30: 61-66.
- Lesage G.D., Schteingart C.D., Hofmann A.F. Effect of bile acid hydrophobicity on biliary transit time and intracellular mobility: a comparison of four fruorescent bile acids analogues. Gastroenterology. 38. 1994; 106(4): Abstr. 929.
- Farrel G.C. Liver disease caused by drugs, anesthetics, and toxins. In: M. Feldman, B.F. Scharschmidt, M.H. Sleisenger, eds. Sleisenger and Fordtran’s Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management. 6th ed. Philadelphia: WB Saunders Company, 1998: 1221-1252.
- Sherlock S., Dooley J. Diseases of the liver and biliary system. 9th ed. Oxford: Blackweel Scientific Publications, 1993. 649 p.
- Lee S.P., Park H.Z., Madani H., Kaler E.W. Partial characterization of a nonmicellar system of cholesterol solubilization in bile. Amer. J. Physiol. 1987; 252: G374-G384.
- Turumin J.L., Shanturov V.A. Pathogenesis and treatment of cholesterol gallstone disease. XIV International Bile Acid Meeting "Bile Acids in Hepatobiliary Diseases - Basic Research and Clinical Application" (Falk Symposium No. 93). Freiburg, Germany, 1996: Abstr 104.
- Jacyna M.R., Ross P.E., Hopwood H., Bouchier I.A.D. Studies on the mechanism of non-visualization of diseased human gallbladders during oral cholecystography. Postgrad Med J 1988; 64(758): 931-935.
- Turumin J.L., Shanturov V.A. The disturbance of the gallbladder bile formation in-patients with cholesterol gallstone disease. XIV International Bile Acid Meeting "Bile Acids in Hepatobiliary Diseases - Basic Research and Clinical Application" (Falk Symposium No. 93). Freiburg, Germany, 1996: Abstr. 105.
- Jazrawi P.P., Pazzi P., Petroni M.L., Northfield T.C. Postprandial refilling and turnover of bile: a novel approach to assessing gallbladder stasis in cholelithiasis. Gastroenterology 1995; 109: 582-591.
- Carey M.C. Formation and growth of cholesterol gallstones: the new synthesis. In: Fromm H., Leuschner U., eds. Bile Acids-Cholestasis-Gallstones. Advances in Basic and Clinical Bile Acid Research. Dordrecht: Kluwer, 1996: 147-175.
- Carey M.C., Cahalane M.J. Whither biliary sludge? Gastroenterology 1988; 95: 508-523.
- Carey M.C., LaMont J.T. Cholesterol gallstone formation. 1. Physical-chemistry of bile and biliary lipid secretion. Prog Liver Dis 1992; 10: 139-163.
- Carey M.C. Pathogenesis of gallstones. Amer J Surg 1993; 165: 410-419.
- Carey M.C. Pathogenesis of cholesterol and pigment gallstones: some radical new concepts. In: Gerok W., Loginov A.S., Pokrowskij V.I., eds. New Trends in Hepatology 1996. Dordrecht: Kluwer, 1996: 64-83.
- LaMont J.T., Carey M.C. Cholesterol gallstone formation. 2. Pathogenesis and pathomechanics. Progr Liver Dis 1992; 10: 165-191.
- Honda A., Yoshida T., Tanaka N., Matsuzaki Y., He B., Shoda J., Osuga T. Increased bile acid concentration in liver tissue with cholesterol gallstone disease. J Gastroenterol 1995; 30(1): 61-66.
- Geraghty J.M., Goldin R.D. Liver changes associated with cholecystitis. J Clin Pathol 1994; 47(5): 457-60.
- Zubovski G.A., ed. Radio and ultrasonic diagnosis of biliary tract diseases. Moscow: Medicine, 1987: 1-240.
- Bayerderffer E; Mannes GA; Richter WO; Ochsenkuhn T; Wiebecke B; Kepcke W; Paumgartner G. Increased serum deoxycholic acid levels in men with colorectal adenomas. Gastroenterology 1993; 104(1): 145-151.
- Bayerderffer E; Mannes GA; Richter WO; Ochsenkuhn T; Seeholzer G; Kepcke W; Wiebecke B; Paumgartner G. Decreased high-density lipoprotein cholesterol and increased low-density cholesterol levels in patients with colorectal adenomas. Ann Intern Med 1993; 118(7): 481-487.
- Bayerderffer E; Mannes GA; Ochsenkuhn T; Dirschedl P; Paumgartner G. Variation of serum bile acids in patients with colorectal adenomas during a one-year follow-up. Digestion 1994; 55(2): 121-129.
- Bayerderffer E; Mannes GA; Ochsenkuhn T; Dirschedl P; Wiebecke B; Paumgartner G. Unconjugated secondary bile acids in the serum of patients with colorectal adenomas. Gut 1995; 36(2): 268-273.
- Ekbom A., Yuen J., Adami H.-O., McLaughlin J.K., Chow W.-H., Persson I., Fraumeni J.F. Cholecystectomy and colorectal cancer. Gastroenterology 1993; 105: 142-147.
- Goldbohm R.A., van den Brandt P.A., van t Veer P., Dorant E., Sturmans F., Hermus R.J. Cholecystectomy and colorectal cancer: evidence from a cohort study on diet and cancer. Int J Cancer 1993; 53(5): 735-739.
- Johansen C., Chow W.-H., Jorgensen T., Mellemkjaer L., Engholm G., Olsen J.H. Risk of colorectal cancer and other cancers in patients with gallstones. Gut 1996; 39: 439-443.
- Chow W.H., Johansen C., Gridley G., Mellemkjair L., Olsen J.H., Fraumeni J.F. Gallstones, cholecystectomy and risk of cancers of the liver, biliary tract and pancreas. Br J Cancer 1999; 79(3-4) 640-644.
- Strom B.L., Soloway R.D., Rios-Dalenz J., Rodriguez-Martinez H.A., West S.L., Kinman J.L., Crowther R.S., Taylor D., Polansky M., Berlin J.A. Biochemical epidemiology of gallbladder cancer. Hepatology 1996, 23: 1402-1411.
- Barthet M., Affriat C., Bernard J.P., Berthezene P., Dagorn J.C., Sahel J. Is biliary lithiasis associated with pancreatographic changes? Gut 1995, 36(5): 761-765.
- Portincasa P., Di Ciaula A., Palmieri V., Velardi A., VanBerge Henegouwen G.P., Palasciano G. Impaired gallbladder and gastric motility and pathological gastro-oesophageal reflux in gallstone patients. Eur J Clin Invest 1997; 27(8): 653-661.
- Stein H.J., Kauer W.K.H., Feussner H., Siewert J.R. Bile acids as components of the duodenogastric refluate: detection, relationship to bilirubin, mechanism of injury, and clinical relevance. Hepatogastroenterology 1999; 46(25): 66-73.
- Fukumoto Y., Murakami F., Andoh M., Mizumachi S., Okita K. Effects of the elevation of serum bile acids on gastric mucosal damage. Hepatol Res 1999; 14(3): 195-203.
|